The power of NAPPA is that by expressing many proteins on a single array, it is possible to test the function of many proteins simultaneously. NAPPA technology is used to explore numerous biological questions including:
Screening for Small Molecule Drug Targets
Characterizing Antibody Specificity and Epitopes
Serum screening to identify antibody immune responses for a variety of diseases
Protein microarrays provide the ideal tool for multiplexed screening of specific antibodies in sera against thousands of different human or viral proteins printed on a standard microscope slide. Below are two examples of autoantibody identification in cancer and infectious disease. For more examples, please visit our research website or review our publications.
Cancer
Cancer patients generate autoantibodies against tumor antigens. These autoantibodies could serve as markers for early cancer detection. Because cancer is not an autoimmune disease per se, the low prevalence and the weak signals of these cancer specific autoantibodies pose greater challenges to the quality of data on our NAPPA platform to identify them as compared with autoantibody biomarker discovery in autoimmune and infectious diseases. Nevertheless, we have successfully applied NAPPA to several cancers’ AAb studies. Most recently, we have applied NAPPA to detect novel tumor antigen-specific AAbs for early breast cancer.
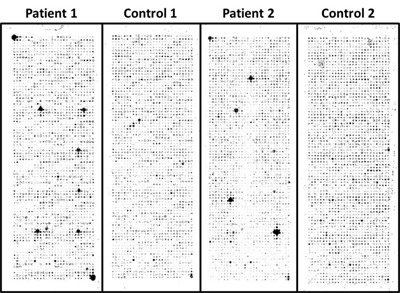 Our study followed three stages: Stage 1, eliminate uninformative antigens; Stage 2, identify breast cancer specific AAbs; Stage 3, blinded validation of AAb. Sera from women undergoing routine screening mammography were selected for a test set for Stage 1 (n=53 cases, 53 controls, Cohort 1), which were screened against nearly 5000 antigens. We removed all but the top 761 antigens from an initial set of 4988 based on ROC methodology. In Stage 2 we identified candidate AAb by printing the selected 761 cDNAs in duplicate on single arrays. These arrays were screened with a separate training set of sera from invasive breast cancer patients (n=51) and from benign breast disease patients (n=39). From these data, 119 antigens were selected as potential biomarkers for further analysis (p<0.05; FDR <13%). In Stage 3 we tested the 119-antigen panel in a blinded independent validation of 51 cases and 38 controls. We tested each antigen using the AUC methods with a focus on highly specific markers. We found evidence (p < 0.05) for 28 of the 119. This represents a statistically significantly higher number of confirmatory findings than would be expected by chance alone (p = 0.0041). For these 28 antigens, we used the threshold that yielded approximately 95% specificity on the training set. In the blinded results, these antigens ranged from 11 – 42% sensitivity with 55 – 100% specificity.
Our study followed three stages: Stage 1, eliminate uninformative antigens; Stage 2, identify breast cancer specific AAbs; Stage 3, blinded validation of AAb. Sera from women undergoing routine screening mammography were selected for a test set for Stage 1 (n=53 cases, 53 controls, Cohort 1), which were screened against nearly 5000 antigens. We removed all but the top 761 antigens from an initial set of 4988 based on ROC methodology. In Stage 2 we identified candidate AAb by printing the selected 761 cDNAs in duplicate on single arrays. These arrays were screened with a separate training set of sera from invasive breast cancer patients (n=51) and from benign breast disease patients (n=39). From these data, 119 antigens were selected as potential biomarkers for further analysis (p<0.05; FDR <13%). In Stage 3 we tested the 119-antigen panel in a blinded independent validation of 51 cases and 38 controls. We tested each antigen using the AUC methods with a focus on highly specific markers. We found evidence (p < 0.05) for 28 of the 119. This represents a statistically significantly higher number of confirmatory findings than would be expected by chance alone (p = 0.0041). For these 28 antigens, we used the threshold that yielded approximately 95% specificity on the training set. In the blinded results, these antigens ranged from 11 – 42% sensitivity with 55 – 100% specificity.
We also used the combined training and validation sets to construct a classifier of patient status. To guard against overfitting, we only used the 28 validated antigens to construct the classifier, and at most 3 antigens were used by each tree in the random forest. The average sensitivity of the classifier was 80.0% and the average specificity was 61.3%.
Infectious Disease
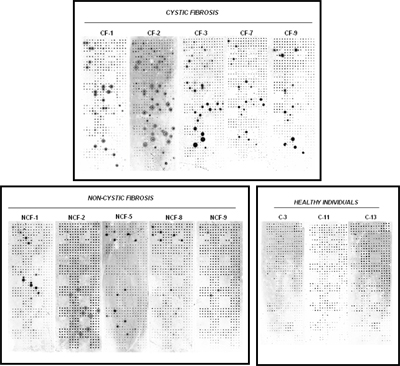
We have extensive experience in working with bacterial and viral pathogen antigens and assaying seroreactivity to these antigens in different diseases. In one such example, we studied Pseudomonas aeruginosa infection, which is responsible for potentially life-threatening infections in individuals with compromised defense mechanisms and those with cystic fibrosis. P. aeruginosa infection is notable for the appearance of a humoral response to some known antigens, such as flagellin C, elastase, alkaline protease, and others. Although a number of immunogenic proteins are known, no effective vaccine has been approved yet. It was reasoned that important immunogens are likely to reside on the surface of the bacterium. Because these outer membrane proteins had extended hydrophobic domains, they were particularly difficult to purify from bacteria, but all of them were readily displayed on the NAPPA arrays. We have reported a comprehensive study of all 262 outer membrane and exported P. aeruginosa PAO1 proteins on NAPPA10. We identified 12 proteins that repeatedly triggered a humoral immune response in cystic fibrosis and acutely infected patients, providing valuable information about which bacterial proteins are frequently recognized by antibodies during the natural course of infection (publication). Similar studies using NAPPA for the detection of immune responses to infectious agents have also been published (VZV) or are underway (cholera, anthrax, tularemia, tuberculosis).
Detect post-translational modifications
Phosphorylation
Protein kinases are important players in all aspects of cell biology, regulating cell signaling pathways that ultimately define the cell fate. The understanding of this key class of proteins has been proven to be fundamental for the treatment and diagnosis of many diseases, especially cancer.Nonetheless, many kinases still have unknown substrates and/or modulators, and a more comprehensive understanding of this class of proteins has the potential to improve disease outcome. The protein array platform is very flexible and many distinct applications can be envisioned, including the screening of new substrates, drug sensitivity and protein/protein interaction, among others.
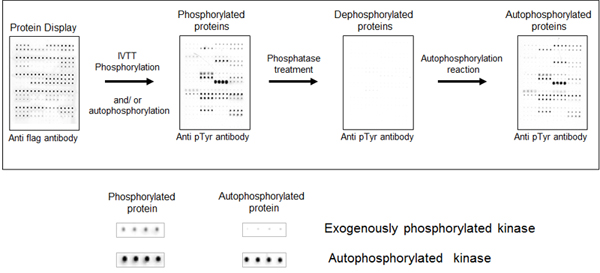
We initially tested the ability to detect protein phosphorylation on the NAPPA array by creating a cloned NAPPA-compatible plasmid collection of all human kinases. Using kinase NAPPA arrays we demonstrated that the kinases on the array display autophosphorylation activity. To demonstrate this, anti-pTyr antibodies detect phosphotyrosines on the kinases on the arrays, which disappear after treatment with phosphatase and then returns after incubation in kinase buffer and ATP (see image). Ongoing experiments are using kinase NAPPA arrays to study the effect of specific kinase inhibitors (small molecules) on the kinase activity and determine the small molecule selectivity and efficacy among many tested kinases, all performed in a single experiment.
AMPylation
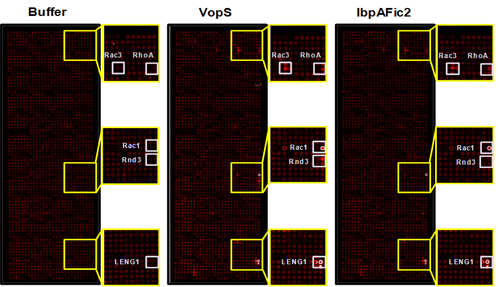 In recent years, a new type of post-translational modification (PTM), AMPylation, has emerged as a fundamental mechanism regulating protein-protein interactions and cell signaling during bacterial pathogenesis. The AMPylation reaction is mediated by virulence factors from bacteria that are secreted into the host cells and can transfer AMP from ATP to the Tyrosine or Threonine group of GTPases. The result of AMPylation is disruption of GTPase binding to downstream effectors such as PAK1 resulting in cell toxicity. AMPylation domains are conserved in both prokaryotic and eukaryotic organisms; therefore, we expect that protein AMPylation plays an important role in a wide range of cellular processes. However, our understanding of AMPylation is limited and identifying new protein AMPylators and their substrates will help illuminate the functional consequences of AMPylation.
In recent years, a new type of post-translational modification (PTM), AMPylation, has emerged as a fundamental mechanism regulating protein-protein interactions and cell signaling during bacterial pathogenesis. The AMPylation reaction is mediated by virulence factors from bacteria that are secreted into the host cells and can transfer AMP from ATP to the Tyrosine or Threonine group of GTPases. The result of AMPylation is disruption of GTPase binding to downstream effectors such as PAK1 resulting in cell toxicity. AMPylation domains are conserved in both prokaryotic and eukaryotic organisms; therefore, we expect that protein AMPylation plays an important role in a wide range of cellular processes. However, our understanding of AMPylation is limited and identifying new protein AMPylators and their substrates will help illuminate the functional consequences of AMPylation.
To address this question, we developed a non-radioactive assay for the detection of AMPylated proteins using NAPPA. With this method we screened 10,000 unique human proteins with two known bacterial AMPylators, VopS and IbpAFic2, and identified new GTPase and non-GTPase substrates dramatically expanding our knowledge of the substrates of these enzymes (see image). These results suggest that this approach could be extended to identify novel substrates of other AMPylators in different species or with different domains.
Identify protein-protein or protein-DNA interactions
 Antibodies are effective tools for biomedical research, disease diagnosis and therapeutics. A thorough understanding of the epitopes antibodies target will improve their utility as research, diagnostic and therapeutic tools.
Antibodies are effective tools for biomedical research, disease diagnosis and therapeutics. A thorough understanding of the epitopes antibodies target will improve their utility as research, diagnostic and therapeutic tools.
Antibodies can be made by immunizing animals with inactivated toxin, bacterial or viral antigens. In the case of bacterial and viral antigens, the entire microorganism is used to immunize the animal. This often produces a robust humoral immune response to immunodominant antigens on the microorganism that can be harnessed to generate either polyclonal or monoclonal antibodies. A major obstacle in using these reagents is identifying the epitope that the antibody binds to on the microorganism. This limitation hampers developing improved affinity reagents and creates a vulnerability in the detection capability of the antibody, should the epitope no longer be present on a select agent. Therefore, a high throughput technology platform to identify protein antigens of antibodies would be highly desirable.
Our Antigen-Specific Epitope-Targeting (ASET) arrays will quickly and easily identify epitopes and enable rapid mapping antibody epitopes at the amino-acid level. Our approach will bypass the expensive cost of synthetic peptides, challenges of antigen purification, and the uncertainty of random peptide library screening. A nested series of deletion mutants, N-terminal (NDM), C-terminal (CDM), internal (IDM) and tiling peptides can be generated rapidly and screened on ASET arrays at a low cost. Hundreds of arrays can be produced in one preparation and used for screening for many different antibodies to the target antigens. We have created an ASET array as proof of concept for p53 protein and a specific monoclonal antibody (see figure). Our ASET arrays will allow epitope mapping in physiological buffers or under denaturing conditions.
Quantify protein binding kinetics in high throughput by coupling NAPPA with surface plasmon resonance (SPRi)
Protein signaling via protein interactions is central to virtually all cellular processes and diseases. Current methods to detect and characterize protein-protein interactions generally fall into two categories: low throughput, which obtain detailed information on protein interactions but operate on only a few proteins at once (e.g., surface plasmon resonance; SPR), or high throughput, which have high false positive and negative rates, and provide little to no quantitative information pertaining to the strength and rate of those interactions (e.g., yeast two hybrid).
To address this issue, our lab has developed a high throughput approach that quantitatively characterizes more than 400 protein interactions simultaneously, collecting information on both interaction affinity and kinetics. In this platform, we have coupled NAPPA with SPR, enabling the assessment of affinities ranging from low micromolarity to high picomolarity. The protein arrays are produced by in situ expression of proteins on the array surface immediately prior to testing.
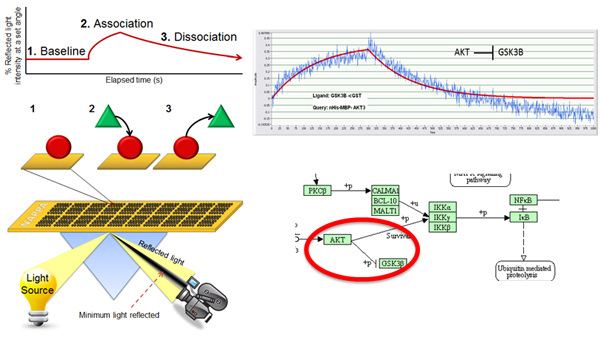
This is a highly reproducible method that displays more consistent levels of protein, costs less and requires much less time than purifying proteins. Virtually all proteins can be produced (including membrane and large proteins) and can be matched to the appropriate expression system (e.g., human proteins made in a human milieu) minutes before the experiment, thus increasing the chance that the proteins are properly folded and functional.
The format has been used to characterize networks of interactions in a signaling pathway, including affinities and kinetics. This open format allows for testing the effects of post translational modifications (PTMs) by testing interactions in their presence and absence. It can also be used to explore the quantitative effects of numerous mutations and isoforms on the ability of a protein to interact with its targets.